45 Illustrations of Thermodynamics
Illustrations of Thermodynamics
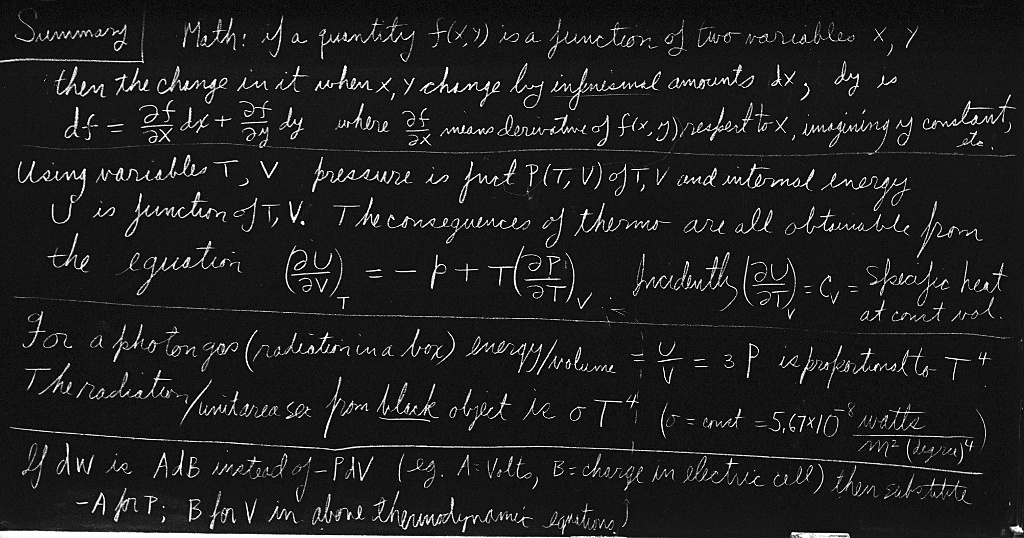
45–1Internal energy
Thermodynamics is a rather difficult and complex subject when we come to apply it, and it is not appropriate for us to go very far into the applications in this course. The subject is of very great importance, of course, to engineers and chemists, and those who are interested in the subject can learn about the applications in physical chemistry or in engineering thermodynamics. There are also good equation reference books, such as Zemansky’s Heat and Thermodynamics, where one can learn more about the subject. In the Encyclopedia Britannica, fourteenth edition, one can find excellent articles on thermodynamics and thermochemistry, and in the article on chemistry, the sections on physical chemistry, vaporization, liquefication of gases, and so on.
The subject of thermodynamics is complicated because there are so many different ways of describing the same thing. If we wish to describe the behavior of a gas, we can say that the pressure depends on the temperature and on the volume, or we can say that the volume depends on the temperature and the pressure. Or with respect to the internal energy $U$, we might say that it depends on the temperature and volume, if those are the variables we have chosen—but we might also say that it depends on the temperature and the pressure, or the pressure and the volume, and so on. In the last chapter we discussed another function of temperature and volume, called the entropy $S$, and we can of course construct as many other functions of these variables as we like: $U - TS$ is a function of temperature and volume. So we have a large number of different quantities which can be functions of many different combinations of variables.
To keep the subject simple in this chapter, we shall decide at the start to use temperature and volume as the independent variables. Chemists use temperature and pressure, because they are easier to measure and control in chemical experiments, but we shall use temperature and volume throughout this chapter, except in one place where we shall see how to make the transformation into the chemists’ system of variables.
We shall first, then, consider only one system of independent variables: temperature and volume. Secondly, we shall discuss only two dependent functions: the internal energy and the pressure. All the other functions can be derived from these, so it is not necessary to discuss them. With these limitations, thermodynamics is still a fairly difficult subject, but it is not quite so impossible!
First we shall review some mathematics. If a quantity is a function of two variables, the idea of the derivative of the quantity requires a little more careful thought than for the case where there is only one variable. What do we mean by the derivative of the pressure with respect to the temperature? The pressure change accompanying a change in the temperature depends partly, of course, on what happens to the volume while $T$ is changing. We must specify the change in $V$ before the concept of a derivative with respect to $T$ has a precise meaning. We might ask, for example, for the rate of change of $P$ with respect to $T$ if $V$ is held constant. This ratio is just the ordinary derivative that we usually write as $dP/dT$. We customarily use a special symbol, $\ddpl{P}{T}$, to remind us that $P$ depends on another variable $V$ as well as on $T$, and that this other variable is held constant. We shall not only use the symbol $\partial$ to call attention to the fact that the other variable is held constant, but we shall also write the variable that is held constant as a subscript, $(\ddpl{P}{T})_V$. Since we have only two independent variables, this notation is redundant, but it will help us keep our wits about us in the thermodynamic jungle of partial derivatives.
Let us suppose that the function $f(x,y)$ depends on the two independent variables $x$ and $y$. By $(\ddpl{f}{x})_y$ we mean simply the ordinary derivative, obtained in the usual way, if we treat $y$ as a constant: \begin{equation*} \biggl(\ddp{f}{x}\biggr)_y = \operatorname*{limit}_{\Delta x \to 0} \frac{f(x + \Delta x,y) - f(x,y)}{\Delta x}. \end{equation*} Similarly, we define \begin{equation*} \biggl(\ddp{f}{y}\biggr)_x = \operatorname*{limit}_{\Delta y \to 0} \frac{f(x, y + \Delta y) - f(x,y)}{\Delta y}. \end{equation*} For example, if $f(x,y) = x^2 + yx$, then $(\ddpl{f}{x})_y = 2x + y$, and $(\ddpl{f}{y})_x = x$. We can extend this idea to higher derivatives: $\partial^2f/\partial y^2$ or $\partial^2f/\partial y\partial x$. The latter symbol indicates that we first differentiate $f$ with respect to $x$, treating $y$ as a constant, then differentiate the result with respect to $y$, treating $x$ as a constant. The actual order of differentiation is immaterial: $\partial^2f/\partial x\partial y = \partial^2f/\partial y\partial x$.
We will need to compute the change $\Delta f$ in $f(x,y)$ when $x$ changes to $x + \Delta x$ and $y$ changes to $y + \Delta y$. We assume throughout the following that $\Delta x$ and $\Delta y$ are infinitesimally small: \begin{align} \Delta f &= f(x + \Delta x, y + \Delta y) - f(x,y)\notag\\[2.5ex] &= \underbrace{f(x + \Delta x, y + \Delta y) - f(x,y + \Delta y)} + \underbrace{f(x,y + \Delta y) - f(x,y)}\notag\\[.5ex] \label{Eq:I:45:1} &= \kern{5em}\displaystyle{\Delta x\biggl(\ddp{f}{x}\biggr)_y} \kern{5em}+ \kern{2.2em}\displaystyle{\Delta y\biggl(\ddp{f}{y}\biggr)_x} \end{align} \begin{alignat}{2} \Delta f &=\phantom{+}f(x + \Delta x, y + \Delta y) &-& \;\; f(x,y)&&\notag\\[2.5ex] &=\phantom{+}f(x + \Delta x, y + \Delta y) &-& \;\; f(x,y + \Delta y)&&\notag\\[1ex] &\phantom{=}+ f(x,y + \Delta y) &-& \;\; f(x,y)&&\notag\\[2.5ex] \label{Eq:I:45:1} &=\Delta x\biggl(\ddp{f}{x}\biggr)_y\!+\Delta y\biggl(\ddp{f}{y}&\biggr)_x& \end{alignat} The last equation is the fundamental relation that expresses $\Delta f$ in terms of $\Delta x$ and $\Delta y$.
As an example of the use of this relation, let us calculate the change in the internal energy $U(T,V)$ when the temperature changes from $T$ to $T + \Delta T$ and the volume changes from $V$ to $V + \Delta V$. Using Eq. (45.1), we write \begin{equation} \label{Eq:I:45:2} \Delta U = \Delta T\biggl(\ddp{U}{T}\biggr)_V + \Delta V\biggl(\ddp{U}{V}\biggr)_T. \end{equation} In our last chapter we found another expression for the change $\Delta U$(* work, $W$ = 압력 곱하기 부피 변화) in the internal energy when a quantity of heat $\Delta Q$ was added to the gas: \begin{equation} \label{Eq:I:45:3} \Delta U = \Delta Q - P\,\Delta V. \end{equation} In comparing Eqs. (45.2) and (45.3) one might at first be inclined to think that $P = -(\ddpl{U}{V})_T$, but this is not correct. To obtain the correct relation, let us first suppose that we add a quantity of heat $\Delta Q$ to the gas while keeping the volume constant, so that $\Delta V = 0$. With $\Delta V = 0$, Eq. (45.3) tells us that $\Delta U = \Delta Q$, and Eq. (45.2) tells us that $\Delta U =(\ddpl{U}{T})_V\,\Delta T$, so that $(\ddpl{U}{T})_V = \Delta Q/\Delta T$. The ratio $\Delta Q/\Delta T$, the amount of heat one must put into a substance in order to change its temperature by one degree with the volume held constant, is called the specific heat at constant volume and is designated by the symbol $C_V$. By this argument we have shown that \begin{equation} \label{Eq:I:45:4} \biggl(\ddp{U}{T}\biggr)_V = C_V. \end{equation}
Now let us again add a quantity of heat $\Delta Q$ to the gas, but this time we will hold $T$ constant and allow the volume to change by $\Delta V$. The analysis in this case is more complex, but we can calculate $\Delta U$ by the argument of Carnot, making use of the Carnot cycle we introduced in the last chapter.

Pressure-volume diagram for a Carnot cycle. The curves marked $T$ and $T - \Delta T$ are isothermal lines; the steeper curves are adiabatic lines. $\Delta V$ is the volume change as heat $\Delta Q$ is added to the gas at constant temperature $T$. $\Delta P$ is the pressure change at constant volume as the gas temperature is changed from $T$ to $T - \Delta T$
The pressure-volume diagram for the Carnot cycle is shown in Fig. 45–1. As we have already shown, the total amount of work done by the gas in a reversible cycle is $\Delta Q(\Delta T/T)$, where $\Delta Q$ is the amount of heat energy added to the gas as it expands isothermally at temperature $T$ from volume $V$ to $V + \Delta V$, and $T - \Delta T$ is the final temperature reached by the gas as it expands adiabatically on the second leg of the cycle.
2022.1.22: By 44.13, $\frac{Q_1}{T_1}=\frac{Q_2}{T_2}, \,W=Q_1-Q_2=Q_1{\frac{T_1-T_2}{T_1}}$, if we let $T_1=T,\, T_2=T-\Delta T,\, Q_1=\Delta Q$, $W= \Delta Q \frac{\Delta T}{T}$
Now we will show that this work done is also given by the shaded area in Fig. 45–1. In any circumstances, the work done by the gas is $\int P\,dV$, and is positive when the gas expands and negative when the gas is compressed. If we plot $P$ vs. $V$, the variation of $P$ and $V$ is represented by a curve which gives the value of $P$ corresponding to a particular value of $V$. As the volume changes from one value to another, the work done by the gas, the integral $\int P\,dV$, is the area under the curve connecting the initial and final values of $V$. When we apply this idea to the Carnot cycle, we see that as we go around the cycle, paying attention to the sign of the work done by the gas, the net work done by the gas is just the shaded area in Fig. 45–1.Now we want to evaluate the shaded area geometrically. The cycle we have used in Fig. 45–1 differs from that used in the previous chapter in that we now suppose that $\Delta T$ and $\Delta Q$ are infinitesimally small. We are working between adiabatic lines and isothermal lines that are very close together, and the figure described by the heavy lines in Fig. 45–1 will approach a parallelogram as the increments $\Delta T$ and $\Delta Q$ approach zero. The area of this parallelogram is just $\Delta V\,\Delta P$, where $\Delta V$ is the change in volume as energy $\Delta Q$ is added to the gas at constant temperature, and $\Delta P$ is the change in pressure as the temperature changes by $\Delta T$ at constant volume. One can easily show that the shaded area in Fig. 45–1 is given by $\Delta V\,\Delta P$ by recognizing that the shaded area is equal to the area enclosed by the dotted lines in Fig. 45–2, which in turn differs from the rectangle bounded by $\Delta P$ and $\Delta V$ only by the addition and subtraction of the equal triangular areas in Fig. 45–2.

Now let us summarize the results of the arguments we have developed so far: \begin{equation} \left.\! \begin{array}{l} \displaystyle\qquad\text{Work done by the gas} = \text{shaded area} = \Delta V\,\Delta P = \Delta Q\biggl(\frac{\Delta T}{T}\biggr)\\ \text{or}\\[1ex] \displaystyle\qquad\frac{\Delta T}{T}\cdot(\text{heat needed to change $V$ by $\Delta V$})_{\text{constant $T$}}\\[1.5ex] \displaystyle\quad = \Delta V\cdot(\text{change in $P$ when $T$ changes by $\Delta T$})_{\text{constant $V$}}\\[2ex] \text{or}\\[.5ex] \displaystyle\qquad\frac{1}{\Delta V}\cdot(\text{heat needed to change $V$ by $\Delta V$})_T = T(\ddpl{P}{T})_V. \end{array}\!\right\} \label{Eq:I:45:5} \end{equation} \begin{equation} \left.\! \begin{array}{l} \quad\begin{gathered} \displaystyle\text{Work done by the gas} = \text{shaded area}\\[1ex] = \Delta V\,\Delta P = \Delta Q\biggl(\frac{\Delta T}{T}\biggr) \end{gathered}\\[-2ex] \text{or}\\[1ex] \displaystyle\quad\frac{\Delta T}{T}\cdot \begin{pmatrix} \text{heat needed}\\[-.5ex] \text{to change $V$}\\[-.5ex] \text{by $\Delta V$} \end{pmatrix}_{\text{constant $T$}}\\[2ex] % ebook remove % ebook insert: \end{pmatrix}_{\text{constant $T$}}\\[5ex] \displaystyle\quad = \Delta V\cdot \begin{pmatrix} \text{change in $P$}\\[-.5ex] \text{when $T$ changes}\\[-.5ex] \text{by $\Delta T$} \end{pmatrix}_{\text{constant $V$}}\\[.5ex] \text{or}\\[.5ex] \displaystyle\quad\frac{1}{\Delta V}\cdot \begin{pmatrix} \text{heat needed}\\[-.5ex] \text{to change $V$}\\[-.5ex] \text{by $\Delta V$} \end{pmatrix}_T \kern{-1.5ex}= T\biggl(\ddp{P}{T}\biggr)_V. \end{array}\!\right\} \label{Eq:I:45:5} \end{equation}
22.1.26: 다른 것을 같은 데, 왜? 하며... 헷갈렸던, work $\int P\, \Delta V, \Delta P \Delta V$를 뛰면서 정리한 후, 든 생각:
generalize function, 복소수, 디랙, 슈레딩거, 사영기하학 등이 우리 3차원 세계는 고차원의 projection이란 생각. 우리가 다루는 방정식들의 고차원 방정식을 찾아낸다면 꿈, 차원 문제도 해결될 거다
Now we can proceed to calculate $(\ddpl{U}{V})_T$. By how much would the internal energy $U$ change if we changed the volume by $\Delta V$? First, $U$ changes because heat is put in, and second, $U$ changes because work is done. The heat put in is \begin{equation*} \Delta Q = T\biggl(\ddp{P}{T}\biggr)_V\Delta V, \end{equation*} according to Eq. (45.5), and the work done on the substance is $-P\,\Delta V$. Therefore the change $\Delta U$ in internal energy has two pieces: \begin{equation} \label{Eq:I:45:6} \Delta U = T\biggl(\ddp{P}{T}\biggr)_V\Delta V - P\,\Delta V. \end{equation} Dividing both sides by $\Delta V$, we find for the rate of change of $U$ with $V$ at constant $T$ \begin{equation} \label{Eq:I:45:7} \biggl(\ddp{U}{V}\biggr)_T = T\biggl(\ddp{P}{T}\biggr)_V - P. \end{equation} In our thermodynamics, in which $T$ and $V$ are the only variables and $P$ and $U$ are the only functions, Eqs. (45.3) and (45.7) are the basic equations from which all the results of the subject can be deduced.
45–2Applications
Now let us discuss the meaning of Eq. (45.7) and see why it answers the questions which we proposed in our last chapter. We considered the following problem: in kinetic theory it is obvious that an increase in temperature leads to an increase in pressure, because of the bombardments of the atoms on a piston. For the same physical reason, when we let the piston move back, heat is taken out of the gas and, in order to keep the temperature constant, heat will have to be put back in. The gas cools when it expands, and the pressure rises when it is heated. There must be some connection between these two phenomena, and this connection is given explicitly in Eq. (45.7). If we hold the volume fixed and increase the temperature, the pressure rises at a rate $(\ddpl{P}{T})_V$. Related to that fact is this: if we increase the volume, the gas will cool unless we pour some heat in to maintain the temperature constant, and $(\ddpl{U}{V})_T$ tells us the amount of heat needed to maintain the temperature. Equation (45.7) expresses the fundamental interrelationship between these two effects. That is what we promised we would find when we came to the laws of thermodynamics. Without knowing the internal mechanism of the gas, and knowing only that we cannot make perpetual motion of the second type, we can deduce the relationship between the amount of heat needed to maintain a constant temperature when the gas expands, and the pressure change when the gas is heated at constant volume!(* 아래: voltage와 temperature의 관계)
Now that we have the result we wanted for a gas, let us consider the rubber band. When we stretch a rubber band, we find that its temperature rises, and when we heat a rubber band, we find that it pulls itself in.
2022.2.2: 에너지 효과 차이는 구조
열을 가하면, 실린더와 물은 부피가 느는 반면, 고무 줄은 줄어든다. 이거는 구조차이라고 밖에 할 수 없다 => internal machinery of rubber
2022.2.21&3.6: $PV=\frac{F}A=F\frac{V}A=Fl$, 에너지를 주입하면 힘, 압력 등이 기체 입자나 전하에 가해져 시스템에 일을 하게 된다는 걸 보여줌
식 (45.8), (45.9)는 formal한 단순 대입이 아니라
주입된 순수 열에너지와 그에 의한 압력 또는 걸린 전하 등에 의한 work(= 힘$\times$거리)로 봐야 한다. Such as...$E$는 단위 에너지, $Z$는 갯수 => $EZ$는 일, 즉 에너지(참조: $\frac{포텐셜 에너지}{갯수}\times {갯수}$)
경제적 상황에 적용한다면, $Q$를 돈, $E$는 사람당 생산하는 돈, $Z$는 사람. 어떤 시점에서의 총자산 변화 $\Delta U$=$\Delta Q$(투자된 돈)+ $E\Delta Z$(이익)
이런 생각흐름을 따르면, 물질은 아주 작은
증기기관과 배터리의 집합체라 할 수 있다는 거.
22.4.28: (45.3), (45.7), (45.9)과 기
기의 흐름이란? 기의 활력 움직임, 수면파처럼. 통증은 신경에 대한 압력이 뇌에 전달되는 것이고 신경에 대한 압박은 혈액 유통이 원활하지 않을 경우 발생. 혈액이 막히거나 몰릴 때 주변 신경을 압박하기 때문. 그 혈액 유통 가능케 하는 것은 심장운동과 실핏줄 역학으로 한의학에서 기 순환이라 부른다. 실핏줄의 원활한 유통은 기의 활동성에 의하니, 한의에서는 기 흐름을 좋게 하기 위한 방법으로 열 가하기(뜸), 뻥뚫어 부항, 침술 사용.
뜸은 열로, 전자침은 전파로 기에 활력을 불어 넣는다(* 통증과 기)
Now we have assumed that the volume of the battery can be maintained constant, since we have omitted the $P\,\Delta V$ term when we set the work done by the battery equal to $E\,\Delta Z$. It turns out that it is technically quite difficult to keep the volume constant. It is much easier to keep the cell at constant atmospheric pressure. For that reason, the chemists do not like any of the equations we have written above: they prefer equations which describe performance under constant pressure. We chose at the beginning of this chapter to use $V$ and $T$ as independent variables. The chemists prefer $P$ and $T$, and we will now consider how the results we have obtained so far can be transformed into the chemists’ system of variables. Remember that in the following treatment confusion can easily set in because we are shifting gears from $T$ and $V$ to $T$ and $P$.
We started in Eq. (45.3) with $\Delta U = \Delta Q - P\,\Delta V$; $P\,\Delta V$ may be replaced by $E\,\Delta Z$ or $A\,\Delta B$. If we could somehow replace the last term, $P\,\Delta V$, by $V\,\Delta P$, then we would have interchanged $V$ and $P$, and the chemists would be happy. Well, a clever man noticed that the differential of the product $PV$ is $d(PV) = P\,dV + V\,dP$, and if he added this equality to Eq. (45.3), he obtained \begin{alignat*}{2} \Delta(PV) &\,= P\,\Delta V &&+ V\,\Delta P\\[.65ex] \Delta U &\,= \Delta Q &&- P\,\Delta V\\ \hline \Delta(U + PV) &\,= \Delta Q &&+ V\,\Delta P \end{alignat*} In order that the result look like Eq. (45.3), we define $U + PV$ to be something new, called the enthalpy, $H$, and we write $\Delta H = \Delta Q + V\,\Delta P$.
Now we are ready to transform our results into chemists’ language with the following rules: $U \to H$, $P \to -V$, $V \to P$. For example, the fundamental relationship that chemists would use instead of Eq. (45.7) is \begin{equation*} \biggl(\ddp{H}{P}\biggr)_T = -T\biggl(\ddp{V}{T}\biggr)_P + V. \end{equation*} It should now be clear how one transforms to the chemists’ variables $T$ and $P$. We now go back to our original variables: for the remainder of this chapter, $T$ and $V$ are the independent variables.
Now let us apply the results we have obtained to a number of physical situations. Consider first the ideal gas. From kinetic theory we know that the internal energy of a gas depends only on the motion of the molecules and the number of molecules. The internal energy depends on $T$, but not on $V$(* 39.9, 39.10 and 39.11). If we change $V$, but keep $T$ constant, $U$ is not changed. Therefore $(\ddpl{U}{V})_T = 0$, and Eq. (45.7) tells us that for an ideal gas \begin{equation} \label{Eq:I:45:10} T\biggl(\ddp{P}{T}\biggr)_V - P = 0. \end{equation} Equation (45.10) is a differential equation that can tell us something about $P$. We take account of the partial derivatives in the following way: Since the partial derivative is at constant $V$, we will replace the partial derivative by an ordinary derivative and write explicitly, to remind us, “constant $V$.” Equation (45.10) then becomes \begin{equation} \label{Eq:I:45:11} T\,\frac{\Delta P}{\Delta T} - P = 0;\quad \text{const $V$}, \end{equation} which we can integrate to get \begin{alignat}{2} \ln P &\;= \ln T + \text{const};\quad &&\text{const $V$},\notag\\[1ex] \label{Eq:I:45:12} P &\;= \text{const}\times T;\quad &&\text{const $V$}. \end{alignat} We know that for an ideal gas the pressure per mole is equal to \begin{equation} \label{Eq:I:45:13} P = \frac{RT}{V}, \end{equation} which is consistent with (45.12), since $V$ and $R$ are constants. Why did we bother to go through this calculation if we already knew the results? Because we have been using two independent definitions of temperature! At one stage we assumed that the kinetic energy of the molecules was proportional to the temperature, an assumption that defines one scale of temperature which we will call the ideal gas scale. The $T$ in Eq. (45.13) is based on the gas scale. We also call temperatures measured on the gas scale kinetic temperatures. Later, we defined the temperature in a second way which was completely independent of any substance(See 44-5). From arguments based on the Second Law we defined what we might call the “grand thermodynamic absolute temperature” $T$, the $T$ that appears in Eq. (45.12). What we proved here is that the pressure of an ideal gas (defined as one for which the internal energy does not depend on the volume) is proportional to the grand thermodynamic absolute temperature. We also know that the pressure is proportional to the temperature measured on the gas scale. Therefore we can deduce that the kinetic temperature is proportional to the “grand thermodynamic absolute temperature.” That means, of course, that if we were sensible we could make two scales agree. In this instance, at least, the two scales have been chosen so that they coincide; the proportionality constant has been chosen to be $1$. Most of the time man chooses trouble for himself, but in this case he made them equal!
45–3The Clausius-Clapeyron equation
The vaporization of a liquid is another application of the results we have derived. Suppose we have some liquid in a cylinder, such that we can compress it by pushing on the piston, and we ask ourselves, “If we keep the temperature constant, how does the pressure vary with volume?” In other words, we want to draw an isothermal line on the $P$-$V$ diagram. The substance in the cylinder is not the ideal gas that we considered earlier; now it may be in the liquid or the vapor phase, or both may be present. If we apply sufficient pressure, the substance will condense to a liquid. Now if we squeeze still harder, the volume changes very little, and our isothermal line rises rapidly with decreasing volume, as shown at the left in Fig. 45–3.

Fig. 45–3.Isothermal lines for a condensable vapor compressed in a cylinder. At the left, the substance is in the liquid phase. At the right, the substance is vaporized. In the center, both liquid and vapor are present in the cylinder.
If we increase the volume by pulling the piston out, the pressure drops until we reach the point at which the liquid starts to boil, and then vapor starts to form. If we pull the piston out farther, all that happens is that more liquid vaporizes. When there is part liquid and part vapor in the cylinder, the two phases are in equilibrium—liquid is evaporating and vapor is condensing at the same rate. If we make more room for the vapor, more vapor is needed to maintain the pressure, so a little more liquid evaporates, but the pressure remains constant. On the flat part of the curve in Fig. 45–3 the pressure does not change, and the value of the pressure here is called the vapor pressure at temperature $T$. As we continue to increase the volume, there comes a time when there is no more liquid to evaporate. At this juncture, if we expand the volume further, the pressure will fall as for an ordinary gas, as shown at the right of the $P$-$V$ diagram. The lower curve in Fig. 45–3 is the isothermal line at a slightly lower temperature $T - \Delta T$. The pressure in the liquid phase is slightly reduced because liquid expands with an increase in temperature (for most substances, but not for water near the freezing point) and, of course, the vapor pressure is lower at the lower temperature.

Fig. 45–4.Pressure-volume diagram for a Carnot cycle with a condensable vapor in the cylinder. At the left, the substance is in the liquid state. A quantity of heat $L$ is added at temperature $T$ to vaporize the liquid. The vapor expands adiabatically as $T$ changes to $T - \Delta T$.
We will now make a cycle out of the two isothermal lines by connecting them (say by adiabatic lines) at both ends of the upper flat section, as shown in Fig. 45–4. We are going to use the argument of Carnot, which tells us that the heat added to the substance in changing it from a liquid to a vapor is related to the work done by the substance as it goes around the cycle. Let us call $L$ the heat needed to vaporize the substance in the cylinder. As in the argument immediately preceding Eq. (45.5), we know that $L(\Delta T/T) = {}$work done by the substance. As before, the work done by the substance is the shaded area, which is approximately $\Delta P(V_G - V_L)$, where $\Delta P$ is the difference in vapor pressure at the two temperatures $T$ and $T - \Delta T$, $V_G$ is the volume of the gas, and $V_L$ is the volume of the liquid, both volumes measured at the vapor pressure at temperature $T$. Setting these two expressions for the area equal, we get $L\,\Delta T/T = \Delta P(V_G - V_L)$, or \begin{equation} \label{Eq:I:45:14} \frac{L}{T(V_G - V_L)} = (\ddpl{P_{\text{vap}}}{T}). \end{equation} Equation (45.14) gives the relationship between the rate of change of vapor pressure with temperature and the amount of heat required to evaporate the liquid. This relationship was deduced by Carnot, but it is called the Clausius-Clapeyron equation.
Now let us compare Eq. (45.14) with the results deduced from kinetic theory. Usually $V_G$ is very much larger than $V_L$. So $V_G - V_L \approx V_G = RT/P$ per mole. If we further assume that $L$ is a constant, independent of temperature—not a very good approximation—then we would have $\ddpl{P}{T} = L/(RT^2/P)$. The solution of this differential equation is \begin{equation} \label{Eq:I:45:15} P = \text{const}\,e^{-L/RT}. \end{equation} Let us compare this with the pressure variation with temperature that we deduced earlier from kinetic theory. Kinetic theory indicated the possibility, at least roughly, that the number of molecules per unit volume of vapor above a liquid would be(* the general rule in two different regions ) \begin{equation} \label{Eq:I:45:16} n = \biggl(\frac{1}{V_a}\biggr)e^{-(U_G - U_L)/RT}, \end{equation} where $U_G - U_L$ is the internal energy per mole in the gas minus the internal energy per mole in the liquid, i.e., the energy needed to vaporize a mole of liquid. Equation (45.15) from thermodynamics and Eq. (45.16) from kinetic theory are very closely related because the pressure is $nkT$, but they are not exactly the same. However, they will turn out to be exactly the same if we assume $U_G - U_L = \text{const}$, instead of $L = \text{const}$. If we assume $U_G - U_L = \text{const}$, independent of temperature, then the argument leading to Eq. (45.15) will produce Eq. (45.16). Since the pressure is constant while the volume is changing, the change in internal energy $U_G-U_L$ is equal to the heat $L$ put in minus the work done $P(V_G-V_L)$, so $L=(U_G+PV_G)-(U_L+PV_L)$.
2022.3.22:
1. 이 부분 수정됨. => 원본
2. (45.14) 식에서 subscript 무시 => $(\ddpl{P}{T})=\frac{L}{T(V_G - V_L)}=\frac{(U_G+PV_G)-(U_L+PV_L)}{T(V_G - V_L)}=\frac{U_G-U_L}{T(V_G-V_L)} +\frac{P}T$. 여기에 $V_G - V_L \approx V_G = RT/P$를 대입.
$(\ddpl{P}{T})=\frac{(U_G-U_L)P}{RT^2} +\frac{P}T$
=> $\frac{dP}P= (\frac{U_G-U_L}{RT^2}+\frac{1}T) dT$, $R$과 $U_G-U_L$이 상수니까, $lnP= -\frac{U_G-U_L}{RT} + lnT+ const$ => $ln{\frac{P}T}= -\frac{U_G-U_L}{RT}+ const$ => $\frac{P}T= \text{const}\, e^{-\frac{U_G-U_L}{RT}}$ =>
$P= \text{const}\,T e^{-\frac{U_G-U_L}{RT}} =nkT$, which we get the desired.
* 5.27: 압력: 규제 등 탄압, 온도: 활동 범위, 부피: 활동 인간 수, 액화: 은퇴, 포기 인간 수, 기체화: 활동 인간 수
This comparison shows the advantages and disadvantages of thermodynamics over kinetic theory: First of all, Eq. (45.14) obtained by thermodynamics is exact, while Eq. (45.16) can only be approximated, for instance, if $U$ is nearly constant, and if the model is right. Second, we may not understand correctly how the gas goes into the liquid; nevertheless, Eq. (45.14) is right, while (45.16) is only approximate. Third, although our treatment applies to a gas condensing into a liquid, the argument is true for any other change of state. For instance, the solid-to-liquid transition has the same kind of curve as that shown in Figs. 45–3 and 45–4. Introducing the latent heat for melting, $M$/mole, the formula analogous to Eq. (45.14) then is $(\ddpl{P_{\text{melt}}}{T})_V = M/[T(V_{\text{liq}} - V_{\text{solid}})]$. Although we may not understand the kinetic theory of the melting process, we nevertheless have a correct equation. However, when we can understand the kinetic theory, we have another advantage. Equation (45.14) is only a differential relationship, and we have no way of obtaining the constants of integration. In the kinetic theory we can obtain the constants also if we have a good model that describes the phenomenon completely. So there are advantages and disadvantages to each. When knowledge is weak and the situation is complicated, thermodynamic relations are really the most powerful. When the situation is very simple and a theoretical analysis can be made, then it is better to try to get more information from theoretical analysis.(참조: 온도의 universality, ideal gas로부터 Q/T)
One more example: blackbody radiation. We have discussed a box containing radiation and nothing else. We have talked about the equilibrium between the oscillator and the radiation. We also found that the photons hitting the wall of the box would exert the pressure $P$, and we found $PV = U/3$, where $U$ is the total energy of all the photons and $V$ is the volume of the box. If we substitute $U = 3PV$ in the basic Eq. (45.7), we find1 \begin{equation} \label{Eq:I:45:17} \biggl(\ddp{U}{V}\biggr)_T = 3P = T\biggl(\ddp{P}{T}\biggr)_V - P. \end{equation} Since the volume of our box is constant, we can replace $(\ddpl{P}{T})_V$ by $dP/dT$ to obtain an ordinary differential equation we can integrate: $\ln P = 4\ln T + \text{const}$, or $P = \text{const}\times T^4$. The pressure of radiation varies as the fourth power of the temperature, and the total energy density of the radiation, $U/V = 3P$, also varies as $T^4$. It is usual to write $U/V = (4\sigma/c)T^4$, where $c$ is the speed of light and $\sigma$ is called the Stefan-Boltzmann constant. It is not possible to get $\sigma$ from thermodynamics alone. Here is a good example of its power, and its limitations. To know that $U/V$ goes as $T^4$ is a great deal, but to know how big $U/V$ actually is at any temperature requires that we go into the kind of detail that only a complete theory can supply. For blackbody radiation we have such a theory and we can derive an expression for the constant $\sigma$ in the following manner.
Let $I(\omega)\,d\omega$ be the intensity distribution, the energy flow through $1$ m² in one second with frequency between $\omega$ and $\omega + d\omega$. The energy density distribution${}={}$energy/volume${}= I(\omega)\,d\omega/c$ is \begin{align*} \frac{U}{V} &= \text{total energy density}\\ &= \int_{\omega = 0}^\infty\text{energy density between $\omega$ and $\omega + d\omega$}\\[1.5ex] &= \int_0^\infty \frac{I(\omega)\,d\omega}{c}. \end{align*} From our earlier discussions(41.16), we know that \begin{equation*} I(\omega) = \frac{\hbar\omega^3}{\pi^2c^2(e^{\hbar\omega/kT} - 1)}. \end{equation*} Substituting this expression for $I(\omega)$ in our equation for $U/V$, we get \begin{equation*} \frac{U}{V} = \frac{1}{\pi^2c^3}\int_0^\infty \frac{\hbar\omega^3\,d\omega}{e^{\hbar\omega/kT} - 1}. \end{equation*} If we substitute $x = \hbar\omega/kT$, the expression becomes \begin{equation*} \frac{U}{V} = \frac{(kT)^4}{\hbar^3\pi^2c^3}\int_0^\infty \frac{x^3\,dx}{e^x - 1}. \end{equation*} This integral is just some number that we can get, approximately, by drawing a curve and taking the area by counting squares. It is roughly $6.5$. The mathematicians among us can show that the integral is exactly $\pi^4/15$.2 Comparing this expression with $U/V = (4\sigma/c)T^4$, we find \begin{equation*} \sigma = \frac{k^4\pi^2}{60\hbar^3c^2} = 5.67\times10^{-8}\,\frac{\text{watts}} {(\text{meter})^2(\text{degree})^4}. \end{equation*}
If we make a small hole in our box, how much energy will flow per second through the hole of unit area? To go from energy density to energy flow, we multiply the energy density $U/V$ by $c$. We also multiply by $\tfrac{1}{4}$, which arises as follows: first, a factor of $\tfrac{1}{2}$, because only the energy which is flowing out escapes; and second, another factor $\tfrac{1}{2}$, because energy which approaches the hole at an angle to the normal is less effective in getting through the hole by a cosine factor. The average value of the cosine factor is $\tfrac{1}{2}$.
2022.3.24: average of $x^2+y^2$, direction an angle θ from it
여러 direction으로부터 구멍을 통해 나가는 걸 계산하는 거니...
unit 두께 표면적 $2\pi$의 half-sphere 에너지, $2\pi$ 중 $\sum_{\theta} {2\pi\sin\theta \Delta \theta \times \cos{\theta}}=\pi \int_0^{\frac{\pi}{2}} \sin{2\theta} d\theta
=-\frac{\pi}2 {\cos{2\theta}}\Biggr|_0^{\frac{\pi}2}=\pi$, 즉 $1/2$
- In this case $(\ddpl{P}{V})_T=0$, because in order to keep the oscillator in equilibrium at a given temperature, the radiation in the neighborhood of the oscillator has to be the same, regardless of the volume of the box. The total quantity of photons inside the box must therefore be proportional to its volume, so the internal energy per unit volume, and thus the pressure, depends only on the temperature. ↩
- Since $(e^x - 1)^{-1} = e^{-x} + e^{-2x} + \dotsb$, the integral is \begin{equation*} \sum_{n=1}^\infty\int_0^\infty e^{-nx}x^3\,dx. \end{equation*} But $\int_0^\infty e^{-nx}\,dx = 1/n$, and differentiating with respect to $n$ three times gives $\int_0^\infty x^3e^{-nx}\,dx = 6/n^4$, so the integral is $6(1 + \tfrac{1}{16} + \tfrac{1}{81} + \dotsb)$ and a good estimate comes from adding the first few terms. In Chapter 50 we will find a way to show that the sum of the reciprocal fourth powers of the integers is, in fact, $\pi^4/90$. ↩