16 The Dependence of Amplitudes on Position
The Dependence of Amplitudes on Position
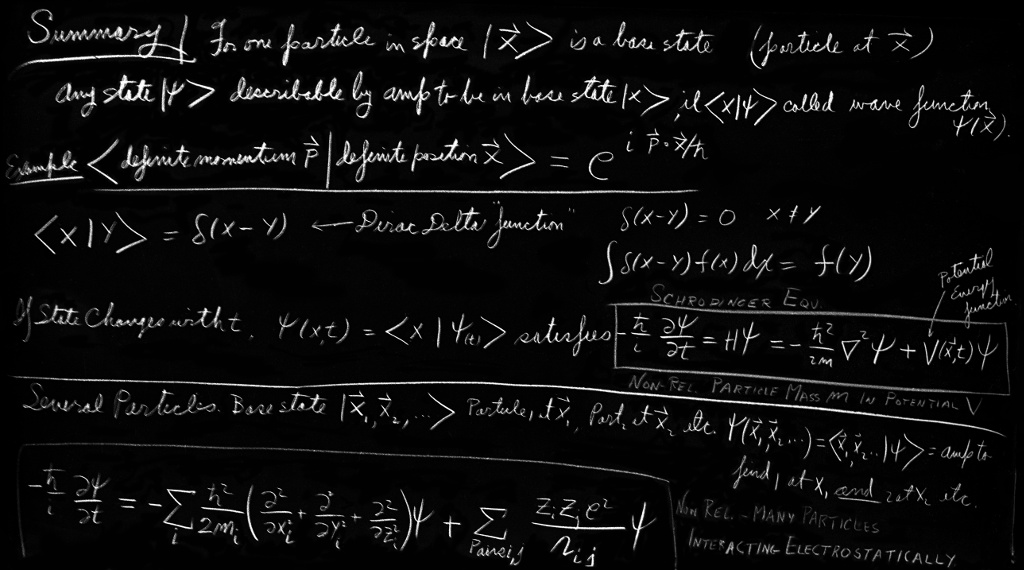
16–1Amplitudes on a line
We are now going to discuss how the probability amplitudes of quantum mechanics vary in space. In some of the earlier chapters you may have had a rather uncomfortable feeling that some things were being left out. For example, when we were talking about the ammonia molecule, we chose to describe it in terms of two base states. For one base state we picked the situation in which the nitrogen atom was “above” the plane of the three hydrogen atoms, and for the other base state we picked the condition in which the nitrogen atom was “below” the plane of the three hydrogen atoms. Why did we pick just these two states? Why is it not possible that the nitrogen atom could be at $2$ angstroms above the plane of the three hydrogen atoms, or at $3$ angstroms, or at $4$ angstroms above the plane? Certainly, there are many positions that the nitrogen atom could occupy. Again when we talked about the hydrogen molecular ion, in which there is one electron shared by two protons, we imagined two base states: one for the electron in the neighborhood of proton number one, and the other for the electron in the neighborhood of proton number two. Clearly we were leaving out many details. The electron is not exactly at proton number two but is only in the neighborhood. It could be somewhere above the proton, somewhere below the proton, somewhere to the left of the proton, or somewhere to the right of the proton.
We intentionally avoided discussing these details. We said that we were interested in only certain features of the problem, so we were imagining that when the electron was in the vicinity of proton number one, it would take up a certain rather definite condition. In that condition the probability to find the electron would have some rather definite distribution around the proton, but we were not interested in the details.
We can also put it another way. In our discussion of a hydrogen molecular ion we chose an approximate description when we described the situation in terms of two base states. In reality there are lots and lots of these states. An electron can take up a condition around a proton in its lowest, or ground, state, but there are also many excited states. For each excited state the distribution of the electron around the proton is different. We ignored these excited states, saying that we were interested in only the conditions of low energy. But it is just these other excited states which give the possibility of various distributions of the electron around the proton. If we want to describe in detail the hydrogen molecular ion, we have to take into account also these other possible base states. We could do this in several ways, and one way is to consider in greater detail states in which the location of the electron in space is more carefully described.
We are now ready to consider a more elaborate procedure which will allow us to talk in detail about the position of the electron, by giving a probability amplitude to find the electron anywhere and everywhere in a given situation. This more complete theory provides the underpinning for the approximations we have been making in our earlier discussions. In a sense, our early equations can be derived as a kind of approximation to the more complete theory.
You may be wondering why we did not begin with the more complete theory and make the approximations as we went along. We have felt that it would be much easier for you to gain an understanding of the basic machinery of quantum mechanics by beginning with the two-state approximations and working gradually up to the more complete theory than to approach the subject the other way around. For this reason our approach to the subject appears to be in the reverse order to the one you will find in many books.
As we go into the subject of this chapter you will notice that we are breaking a rule we have always followed in the past. Whenever we have taken up any subject we have always tried to give a more or less complete description of the physics—showing you as much as we could about where the ideas led to. We have tried to describe the general consequences of a theory as well as describing some specific detail so that you could see where the theory would lead. We are now going to break that rule; we are going to describe how one can talk about probability amplitudes in space and show you the differential equations which they satisfy. We will not have time to go on and discuss many of the obvious implications which come out of the theory. Indeed we will not even be able to get far enough to relate this theory to some of the approximate formulations we have used earlier—for example, to the hydrogen molecule or to the ammonia molecule. For once, we must leave our business unfinished and open-ended. We are approaching the end of our course, and we must satisfy ourselves with trying to give you an introduction to the general ideas and with indicating the connections between what we have been describing and some of the other ways of approaching the subject of quantum mechanics. We hope to give you enough of an idea that you can go off by yourself and by reading books learn about many of the implications of the equations we are going to describe. We must, after all, leave something for the future.
Let’s review once more what we have found out about how an electron can move along a line of atoms. When an electron has an amplitude to jump from one atom to the next, there are definite energy states in which the probability amplitude for finding the electron is distributed along the lattice in the form of a traveling wave. For long wavelengths—for small values of the wave number $k$—the energy of the state is proportional to the square of the wave number. For a crystal lattice with the spacing $b$, in which the amplitude per unit time for the electron to jump from one atom to the next is $iA/\hbar$, the energy of the state is related to $k$ (for small $kb$) by \begin{equation} \label{Eq:III:16:1} E=Ak^2b^2 \end{equation} (see Section 13–2). We also saw that groups of such waves with similar energies would make up a wave packet which would behave like a classical particle with a mass $m_{\text{eff}}$ given by: \begin{equation} \label{Eq:III:16:2} m_{\text{eff}}=\frac{\hbar^2}{2Ab^2}. \end{equation}
Since waves of probability amplitude in a crystal behave like a particle, one might well expect that the general quantum mechanical description of a particle would show the same kind of wave behavior we observed for the lattice. Suppose we were to think of a lattice on a line and imagine that the lattice spacing $b$ were to be made smaller and smaller. In the limit we would be thinking of a case in which the electron could be anywhere along the line. We would have gone over to a continuous distribution of probability amplitudes. We would have the amplitude to find an electron anywhere along the line. This would be one way to describe the motion of an electron in a vacuum. In other words, if we imagine that space can be labeled by an infinity of points all very close together and we can work out the equations that relate the amplitudes at one point to the amplitudes at neighboring points, we will have the quantum mechanical laws of motion of an electron in space.
Let’s begin by recalling some of the general principles of quantum mechanics. Suppose we have a particle which can exist in various conditions in a quantum mechanical system. Any particular condition an electron can be found in, we call a “state,” which we label with a state vector, say $\ket{\phi}$. Some other condition would be labeled with another state vector, say $\ket{\psi}$. We then introduce the idea of base states. We say that there is a set of states $\ket{1}$, $\ket{2}$, $\ket{3}$, $\ket{4}$, and so on, which have the following properties. First, all of these states are quite distinct—we say they are orthogonal. By this we mean that for any two of the base states $\ket{i}$ and $\ket{j}$ the amplitude $\braket{i}{j}$ that an electron known to be in the state $\ket{i}$ is also in the state $\ket{j}$ is equal to zero—unless, of course, $\ket{i}$ and $\ket{j}$ stand for the same state. We represent this symbolically by \begin{equation} \label{Eq:III:16:3} \braket{i}{j}=\delta_{ij}. \end{equation} You will remember that $\delta_{ij}=0$ if $i$ and $j$ are different, and $\delta_{ij}=1$ if $i$ and $j$ are the same number.
Second, the base states $\ket{i}$ must be a complete set, so that any state at all can be described in terms of them. That is, any state $\ket{\phi}$ at all can be described completely by giving all of the amplitudes $\braket{i}{\phi}$ that a particle in the state $\ket{\phi}$ will also be found in the state $\ket{i}$. In fact, the state vector $\ket{\phi}$ is equal to the sum of the base states each multiplied by a coefficient which is the amplitude that the state $\ket{\phi}$ is also in the state $\ket{i}$: \begin{equation} \label{Eq:III:16:4} \ket{\phi}=\sum_i\ket{i}\braket{i}{\phi}. \end{equation}
Finally, if we consider any two states $\ket{\phi}$ and $\ket{\psi}$, the amplitude that the state $\ket{\psi}$ will also be in the state $\ket{\phi}$ can be found by first projecting the state $\ket{\psi}$ into the base states and then projecting from each base state into the state $\ket{\phi}$. We write that in the following way: \begin{equation} \label{Eq:III:16:5} \braket{\phi}{\psi}=\sum_i\braket{\phi}{i}\braket{i}{\psi}. \end{equation} The summation is, of course, to be carried out over the whole set of base states $\ket{i}$.
In Chapter 13 when we were working out what happens with an electron placed on a linear array of atoms, we chose a set of base states in which the electron was localized at one or other of the atoms in the line. The base state $\ket{n}$ represented the condition in which the electron was localized at atom number “$n$.” (There is, of course, no significance to the fact that we called our base states $\ket{n}$ instead of $\ket{i}$.) A little later, we found it convenient to label the base states by the coordinate $x_n$ of the atom rather than by the number of the atom in the array. The state $\ket{x_n}$ is just another way of writing the state $\ket{n}$. Then, following the general rules, any state at all, say $\ket{\psi}$ is described by giving the amplitudes that an electron in the state $\ket{\psi}$ is also in one of the states $\ket{x_n}$. For convenience we have chosen to let the symbol $C_n$ stand for these amplitudes, \begin{equation} \label{Eq:III:16:6} C_n=\braket{x_n}{\psi}. \end{equation}
Since the base states are associated with a location along the line, we can think of the amplitude $C_n$ as a function of the coordinate $x$ and write it as $C(x_n)$. The amplitudes $C(x_n)$ will, in general, vary with time and are, therefore, also functions of $t$. We will not generally bother to show explicitly this dependence.
In Chapter 13 we then proposed that the amplitudes $C(x_n)$ should vary with time in a way described by the Hamiltonian equation (Eq. 13.3). In our new notation this equation is \begin{equation} \label{Eq:III:16:7} i\hbar\ddp{C(x_n)}{t}\!=\!E_0C(x_n)\!-\! AC(x_n\!+\!b)\!-\!AC(x_n\!-\!b). \end{equation} The last two terms on the right-hand side represent the process in which an electron at atom $(n+1)$ or at atom $(n-1)$ can feed into atom $n$.
We found that Eq. (16.7) has solutions corresponding to definite energy states, which we wrote as \begin{equation} \label{Eq:III:16:8} C(x_n)=e^{-iEt/\hbar}e^{ikx_n}. \end{equation} For the low-energy states the wavelengths are large ($k$ is small), and the energy is related to $k$ by \begin{equation} \label{Eq:III:16:9} E=(E_0-2A)+Ak^2b^2, \end{equation} or, choosing our zero of energy so that $(E_0-2A)=0$, the energy is given by Eq. (16.1).
Let’s see what might happen if we were to let the lattice spacing $b$ go to zero, keeping the wave number $k$ fixed. If that is all that were to happen the last term in Eq. (16.9) would just go to zero and there would be no physics. But suppose $A$ and $b$ are varied together so that as $b$ goes to zero the product $Ab^2$ is kept constant1—using Eq. (16.2) we will write $Ab^2$ as the constant $\hbar^2/2m_{\text{eff}}$. Under these circumstances, Eq. (16.9) would be unchanged, but what would happen to the differential equation (16.7)?
First we will rewrite Eq. (16.7) as \begin{equation} \label{Eq:III:16:10} i\hbar\,\ddp{C(x_n)}{t}=(E_0-2A)C(x_n)+A[2C(x_n)- C(x_n+b)-C(x_n-b)]. \end{equation} For our choice of $E_0$, the first term drops out. Next, we can think of a continuous function $C(x)$ that goes smoothly through the proper values $C(x_n)$ at each $x_n$. As the spacing $b$ goes to zero, the points $x_n$ get closer and closer together, and (if we keep the variation of $C(x)$ fairly smooth) the quantity in the brackets is just proportional to the second derivative of $C(x)$. We can write—as you can see by making a Taylor expansion of each term—the equality \begin{equation} \label{Eq:III:16:11} 2C(x)-C(x+b)-C(x-b)\approx-b^2\, \frac{\partial^2C(x)}{\partial x^2}. \end{equation} In the limit, then, as $b$ goes to zero, keeping $b^2A$ equal to $\hbar^2/2m_{\text{eff}}$, Eq. (16.7) goes over into \begin{equation} \label{Eq:III:16:12} i\hbar\,\ddp{C(x)}{t}=-\frac{\hbar^2}{2m_{\text{eff}}}\, \frac{\partial^2C(x)}{\partial x^2}. \end{equation} We have an equation which says that the time rate of change of $C(x)$—the amplitude to find the electron at $x$—depends on the amplitude to find the electron at nearby points in a way which is proportional to the second derivative of the amplitude with respect to position.
The correct quantum mechanical equation for the motion of an electron in free space was first discovered by Schrödinger. For motion along a line it has exactly the form of Eq. (16.12) if we replace $m_{\text{eff}}$ by $m$, the free-space mass of the electron. For motion along a line in free space the Schrödinger equation is \begin{equation} \label{Eq:III:16:13} i\hbar\,\ddp{C(x)}{t}=-\frac{\hbar^2}{2m}\, \frac{\partial^2C(x)}{\partial x^2}. \end{equation}
We do not intend to have you think we have derived the Schrödinger equation but only wish to show you one way of thinking about it. When Schrödinger first wrote it down, he gave a kind of derivation based on some heuristic arguments and some brilliant intuitive guesses. Some of the arguments he used were even false, but that does not matter; the only important thing is that the ultimate equation gives a correct description of nature. The purpose of our discussion is then simply to show you that the correct fundamental quantum mechanical equation (16.13) has the same form you get for the limiting case of an electron moving along a line of atoms. This means that we can think of the differential equation in (16.13) as describing the diffusion of a probability amplitude from one point to the next along the line. That is, if an electron has a certain amplitude to be at one point, it will, a little time later, have some amplitude to be at neighboring points. In fact, the equation looks something like the diffusion equations which we have used in Volume I. But there is one main difference: the imaginary coefficient in front of the time derivative makes the behavior completely different from the ordinary diffusion such as you would have for a gas spreading out along a thin tube. Ordinary diffusion gives rise to real exponential solutions, whereas the solutions of Eq. (16.13) are complex waves.
16–2The wave function
Now that you have some idea about how things are going to look, we want to go back to the beginning and study the problem of describing the motion of an electron along a line without having to consider states connected with atoms on a lattice. We want to go back to the beginning and see what ideas we have to use if we want to describe the motion of a free particle in space. Since we are interested in the behavior of a particle, along a continuum, we will be dealing with an infinite number of possible states and, as you will see, the ideas we have developed for dealing with a finite number of states will need some technical modifications.
We begin by letting the state vector $\ket{x}$ stand for a state in which a particle is located precisely at the coordinate $x$. For every value $x$ along the line—for instance $1.73$, or $9.67$, or $10.00$—there is the corresponding state. We will take these states $\ket{x}$ as our base states and, if we include all the points on the line, we will have a complete set for motion in one dimension. Now suppose we have a different kind of a state, say $\ket{\psi}$, in which an electron is distributed in some way along the line. One way of describing this state is to give all the amplitudes that the electron will be also found in each of the base states $\ket{x}$. We must give an infinite set of amplitudes, one for each value of $x$. We will write these amplitudes as $\braket{x}{\psi}$. Each of these amplitudes is a complex number and since there is one such complex number for each value of $x$, the amplitude $\braket{x}{\psi}$ is indeed just a function of $x$. We will also write it as $C(x)$, \begin{equation} \label{Eq:III:16:14} C(x)\equiv\braket{x}{\psi}. \end{equation}
We have already considered such amplitudes which vary in a continuous way with the coordinates when we talked about the variations of amplitude with time in Chapter 7. We showed there, for example, that a particle with a definite momentum should be expected to have a particular variation of its amplitude in space. If a particle has a definite momentum $p$ and a corresponding definite energy $E$, the amplitude to be found at any position $x$ would look like \begin{equation} \label{Eq:III:16:15} \braket{x}{\psi}=C(x)\propto e^{+ipx/\hbar}. \end{equation} This equation expresses an important general principle of quantum mechanics which connects the base states corresponding to different positions in space to another system of base states—all the states of definite momentum. The definite momentum states are often more convenient than the states in $x$ for certain kinds of problems. Either set of base states is, of course, equally acceptable for a description of a quantum mechanical situation. We will come back later to the matter of the connection between them. For the moment we want to stick to our discussion of a description in terms of the states $\ket{x}$.
Before proceeding, we want to make one small change in notation which we hope will not be too confusing. The function $C(x)$, defined in Eq. (16.14), will of course have a form which depends on the particular state $\ket{\psi}$ under consideration. We should indicate that in some way. We could, for example, specify which function $C(x)$ we are talking about by a subscript say, $C_\psi(x)$. Although this would be a perfectly satisfactory notation, it is a little bit cumbersome and is not the one you will find in most books. Most people simply omit the letter $C$ and use the symbol $\psi$ to define the function \begin{equation} \label{Eq:III:16:16} \psi(x)\equiv C_\psi(x)=\braket{x}{\psi}. \end{equation} Since this is the notation used by everybody else in the world, you might as well get used to it so that you will not be frightened when you come across it somewhere else. Remember though, that we will now be using $\psi$ in two different ways. In Eq. (16.14), $\psi$ stands for a label we have given to a particular physical state of the electron. On the left-hand side of Eq. (16.16), on the other hand, the symbol $\psi$ is used to define a mathematical function of $x$ which is equal to the amplitude to be associated with each point $x$ along the line. We hope it will not be too confusing once you get accustomed to the idea. Incidentally, the function $\psi(x)$ is usually called “the wave function”—because it more often than not has the form of a complex wave in its variables.
Since we have defined $\psi(x)$ to be the amplitude that an electron in the state $\psi$ will be found at the location $x$, we would like to interpret the absolute square of $\psi$ to be the probability of finding an electron at the position $x$. Unfortunately, the probability of finding a particle exactly at any particular point is zero. The electron will, in general, be smeared out in a certain region of the line, and since, in any small piece of the line, there are an infinite number of points, the probability that it will be at any one of them cannot be a finite number. We can only describe the probability of finding an electron in terms of a probability distribution2 which gives the relative probability of finding the electron at various approximate locations along the line. Let’s let $\prob(x,\Delta x)$ stand for the chance of finding the electron in a small interval $\Delta x$ located near $x$. If we go to a small enough scale in any physical situation, the probability will be varying smoothly from place to place, and the probability of finding the electron in any small finite line segment $\Delta x$ will be proportional to $\Delta x$. We can modify our definitions to take this into account.
We can think of the amplitude $\braket{x}{\psi}$ as representing a kind of “amplitude density” for all the base states $\ket{x}$ in a small region. Since the probability of finding an electron in a small interval $\Delta x$ at $x$ should be proportional to the interval $\Delta x$, we choose our definition of $\braket{x}{\psi}$ so that the following relation holds: \begin{equation*} \prob(x,\Delta x)=\abs{\braket{x}{\psi}}^2\,\Delta x. \end{equation*} The amplitude $\braket{x}{\psi}$ is therefore proportional to the amplitude that an electron in the state $\psi$ will be found in the base state $x$ and the constant of proportionality is chosen so that the absolute square of the amplitude $\braket{x}{\psi}$ gives the probability density of finding an electron in any small region. We can write, equivalently, \begin{equation} \label{Eq:III:16:17} \prob(x,\Delta x)=\abs{\psi(x)}^2\,\Delta x. \end{equation}
We will now have to modify some of our earlier equations to make them compatible with this new definition of a probability amplitude. Suppose we have an electron in the state $\ket{\psi}$ and we want to know the amplitude for finding it in a different state $\ket{\phi}$ which may correspond to a different spread-out condition of the electron. When we were talking about a finite set of discrete states, we would have used Eq. (16.5). Before modifying our definition of the amplitudes we would have written \begin{equation} \label{Eq:III:16:18} \braket{\phi}{\psi}=\sum_{\text{all $x$}} \braket{\phi}{x}\braket{x}{\psi}. \end{equation} Now if both of these amplitudes are normalized in the same way as we have described above, then a sum of all the states in a small region of $x$ would be equivalent to multiplying by $\Delta x$, and the sum over all values of $x$ simply becomes an integral. With our modified definitions, the correct form becomes \begin{equation} \label{Eq:III:16:19} \braket{\phi}{\psi}=\int_{\text{all $x$}} \braket{\phi}{x}\braket{x}{\psi}\,dx. \end{equation}
The amplitude $\braket{x}{\psi}$ is what we are now calling $\psi(x)$ and, in a similar way, we will choose to let the amplitude $\braket{x}{\phi}$ be represented by $\phi(x)$. Remembering that $\braket{\phi}{x}$ is the complex conjugate of $\braket{x}{\phi}$, we can write Eq. (16.19) as \begin{equation} \label{Eq:III:16:20} \braket{\phi}{\psi}=\int\phi\cconj(x)\psi(x)\,dx. \end{equation} With our new definitions everything follows with the same formulas as before if you always replace a summation sign by an integral over $x$.
We should mention one qualification to what we have been saying. Any suitable set of base states must be complete if it is to be used for an adequate description of what is going on. For an electron in one dimension it is not really sufficient to specify only the base states $\ket{x}$, because for each of these states the electron may have a spin which is either up or down. One way of getting a complete set is to take two sets of states in $x$, one for up spin and the other for down spin. We will, however, not worry about such complications for the time being.
16–3States of definite momentum
Suppose we have an electron in a state $\ket{\psi}$ which is described by the probability amplitude $\braket{x}{\psi}=\psi(x)$. We know that this represents a state in which the electron is spread out along the line in a certain distribution so that the probability of finding the electron in a small interval $dx$ at the location $x$ is just \begin{equation*} \prob(x,dx)=\abs{\psi(x)}^2\,dx. \end{equation*} What can we say about the momentum of this electron? We might ask what is the probability that this electron has the momentum $p$? Let’s start out by calculating the amplitude that the state $\ket{\psi}$ is in another state $\ket{\mom p}$ which we define to be a state with the definite momentum $p$. We can find this amplitude by using our basic equation for the resolution of amplitudes, Eq. (16.19). In terms of the state $\ket{\mom p}$ \begin{equation} \label{Eq:III:16:21} \braket{\mom p}{\psi}=\int_{x=-\infty}^{+\infty} \braket{\mom p}{x}\braket{x}{\psi}\,dx. \end{equation} And the probability that the electron will be found with the momentum $p$ should be given in terms of the absolute square of this amplitude. We have again, however, a small problem about the normalizations. In general we can only ask about the probability of finding an electron with a momentum in a small range $dp$ at the momentum $p$. The probability that the momentum is exactly some value $p$ must be zero (unless the state $\ket{\psi}$ happens to be a state of definite momentum). Only if we ask for the probability of finding the momentum in a small range $dp$ at the momentum $p$ will we get a finite probability. There are several ways the normalizations can be adjusted. We will choose one of them which we think to be the most convenient, although that may not be apparent to you just now.
We take our normalizations so that the probability is related to the amplitude by \begin{equation} \label{Eq:III:16:22} \prob(p,dp)=\abs{\braket{\mom p}{\psi}}^2\,\frac{dp}{2\pi\hbar}. \end{equation} With this definition the normalization of the amplitude $\braket{\mom p}{x}$ is determined. The amplitude $\braket{\mom p}{x}$ is, of course, just the complex conjugate of the amplitude $\braket{x}{\mom p}$, which is just the one we have written down in Eq. (16.15). With the normalization we have chosen, it turns out that the proper constant of proportionality in front of the exponential is just $1$. Namely, \begin{equation} \label{Eq:III:16:23} \braket{\mom p}{x}=\braket{x}{\mom p}\cconj=e^{-ipx/\hbar}. \end{equation} Equation (16.21) then becomes \begin{equation} \label{Eq:III:16:24} \braket{\mom p}{\psi}=\int_{-\infty}^{+\infty} e^{-ipx/\hbar}\braket{x}{\psi}\,dx. \end{equation} This equation together with Eq. (16.22) allows us to find the momentum distribution for any state $\ket{\psi}$.
Let’s look at a particular example—for instance one in which an electron is localized in a certain region around $x=0$. Suppose we take a wave function which has the following form: \begin{equation} \label{Eq:III:16:25} \psi(x)=Ke^{-x^2/4\sigma^2}. \end{equation} The probability distribution in $x$ for this wave function is the absolute square, or \begin{equation} \label{Eq:III:16:26} \prob(x,dx)=P(x)\,dx=K^2e^{-x^2/2\sigma^2}\,dx. \end{equation}
The probability density function $P(x)$ is the Gaussian curve shown in Fig. 16–1. Most of the probability is concentrated between $x=+\sigma$ and $x=-\sigma$. We say that the “half-width” of the curve is $\sigma$. (More precisely, $\sigma$ is equal to the root-mean-square of the coordinate $x$ for something spread out according to this distribution.) We would normally choose the constant $K$ so that the probability density $P(x)$ is not merely proportional to the probability per unit length in $x$ of finding the electron, but has a scale such that $P(x)\,\Delta x$ is equal to the probability of finding the electron in $\Delta x$ near $x$. The constant $K$ which does this can be found by requiring that $\int_{-\infty}^{+\infty}P(x)\,dx=1$, since there must be unit probability that the electron is found somewhere. Here, we get that $K=(2\pi\sigma^2)^{-1/4}$. [We have used the fact that $\int_{-\infty}^{+\infty}e^{-t^2}\,dt=\sqrt{\pi}$; see Vol. I, footnote 40-1.]
Now let’s find the distribution in momentum. Let’s let $\phi(p)$ stand for the amplitude to find the electron with the momentum $p$, \begin{equation} \label{Eq:III:16:27} \phi(p)\equiv\braket{\mom p}{\psi}. \end{equation} Substituting Eq. (16.25) into Eq. (16.24) we get \begin{equation} \label{Eq:III:16:28} \phi(p)=\int_{-\infty}^{+\infty} e^{-ipx/\hbar}\cdot Ke^{-x^2/4\sigma^2}\,dx. \end{equation} The integral can also be rewritten as \begin{equation} \label{Eq:III:16:29} Ke^{-p^2\sigma^2/\hbar^2}\int_{-\infty}^{+\infty} e^{-(1/4\sigma^2)(x+2ip\sigma^2/\hbar)^2}\,dx. \end{equation} We can now make the substitution $u=x+2ip\sigma^2/\hbar$, and the integral is \begin{equation} \label{Eq:III:16:30} \int_{-\infty}^{+\infty}e^{-u^2/4\sigma^2}\,du=2\sigma\sqrt{\pi}. \end{equation} (The mathematicians would probably object to the way we got there, but the result is, nevertheless, correct.) \begin{equation} \label{Eq:III:16:31} \phi(p)=(8\pi\sigma^2)^{1/4}e^{-p^2\sigma^2/\hbar^2}. \end{equation}
We have the interesting result that the amplitude function in $p$ has precisely the same mathematical form as the amplitude function in $x$; only the width of the Gaussian is different. We can write this as \begin{equation} \label{Eq:III:16:32} \phi(p)=(\eta^2/2\pi\hbar^2)^{-1/4}e^{-p^2/4\eta^2}, \end{equation} where the half-width $\eta$ of the $p$-distribution function is related to the half-width $\sigma$ of the $x$-distribution by \begin{equation} \label{Eq:III:16:33} \eta=\frac{\hbar}{2\sigma}. \end{equation}
Our result says: if we make the width of the distribution in $x$ very small by making $\sigma$ small, $\eta$ becomes large and the distribution in $p$ is very much spread out. Or, conversely: if we have a narrow distribution in $p$, it must correspond to a spread-out distribution in $x$. We can, if we like, consider $\eta$ and $\sigma$ to be some measure of the uncertainty in the localization of the momentum and of the position of the electron in the state we are studying. If we call them $\Delta p$ and $\Delta x$ respectively Eq. (16.33) becomes \begin{equation} \label{Eq:III:16:34} \Delta p\,\Delta x=\frac{\hbar}{2}. \end{equation}
Interestingly enough, it is possible to prove that for any other form of a distribution in $x$ or in $p$, the product $\Delta p\,\Delta x$ cannot be smaller than the one we have found here. The Gaussian distribution gives the smallest possible value for the product of the root-mean-square widths. In general, we can say \begin{equation} \label{Eq:III:16:35} \Delta p\,\Delta x\geq\frac{\hbar}{2}. \end{equation} This is a quantitative statement of the Heisenberg uncertainty principle, which we have discussed qualitatively many times before. We have usually made the approximate statement that the minimum value of the product $\Delta p\,\Delta x$ is of the same order as $\hbar$.
16–4Normalization of the states in $\boldsymbol{x}$
We return now to the discussion of the modifications of our basic equations which are required when we are dealing with a continuum of base states. When we have a finite number of discrete states, a fundamental condition which must be satisfied by the set of base states is \begin{equation} \label{Eq:III:16:36} \braket{i}{j}=\delta_{ij}. \end{equation} If a particle is in one base state, the amplitude to be in another base state is $0$. By choosing a suitable normalization, we have defined the amplitude $\braket{i}{i}$ to be $1$. These two conditions are described by Eq. (16.36). We want now to see how this relation must be modified when we use the base states $\ket{x}$ of a particle on a line. If the particle is known to be in one of the base states $\ket{x}$, what is the amplitude that it will be in another base state $\ket{x'}$? If $x$ and $x'$ are two different locations along the line, then the amplitude $\braket{x}{x'}$ is certainly $0$, so that is consistent with Eq. (16.36). But if $x$ and $x'$ are equal, the amplitude $\braket{x}{x'}$ will not be $1$, because of the same old normalization problem. To see how we have to patch things up, we go back to Eq. (16.19), and apply this equation to the special case in which the state $\ket{\phi}$ is just the base state $\ket{x'}$. We would have then \begin{equation} \label{Eq:III:16:37} \braket{x'}{\psi}=\int\braket{x'}{x}\psi(x)\,dx. \end{equation} Now the amplitude $\braket{x}{\psi}$ is just what we have been calling the function $\psi(x)$. Similarly the amplitude $\braket{x'}{\psi}$, since it refers to the same state $\ket{\psi}$, is the same function of the variable $x'$, namely $\psi(x')$. We can, therefore, rewrite Eq. (16.37) as \begin{equation} \label{Eq:III:16:38} \psi(x')=\int\braket{x'}{x}\psi(x)\,dx. \end{equation} This equation must be true for any state $\ket{\psi}$ and, therefore, for any arbitrary function $\psi(x)$. This requirement should completely determine the nature of the amplitude $\braket{x}{x'}$—which is, of course, just a function that depends on $x$ and $x'$.
Our problem now is to find a function $f(x,x')$, which when multiplied into $\psi(x)$, and integrated over all $x$ gives just the quantity $\psi(x')$. It turns out that there is no mathematical function which will do this! At least nothing like what we ordinarily mean by a “function.”
Suppose we pick $x'$ to be the special number $0$ and define the amplitude $\braket{0}{x}$ to be some function of $x$, let’s say $f(x)$. Then Eq. (16.38) would read as follows: \begin{equation} \label{Eq:III:16:39} \psi(0)=\int f(x)\psi(x)\,dx. \end{equation} What kind of function $f(x)$ could possibly satisfy this equation? Since the integral must not depend on what values $\psi(x)$ takes for values of $x$ other than $0$, $f(x)$ must clearly be $0$ for all values of $x$ except $0$. But if $f(x)$ is $0$ everywhere, the integral will be $0$, too, and Eq. (16.39) will not be satisfied. So we have an impossible situation: we wish a function to be $0$ everywhere but at a point, and still to give a finite integral. Since we can’t find a function that does this, the easiest way out is just to say that the function $f(x)$ is defined by Eq. (16.39). Namely, $f(x)$ is that function which makes (16.39) correct. The function which does this was first invented by Dirac and carries his name. We write it $\delta(x)$. All we are saying is that the function $\delta(x)$ has the strange property that if it is substituted for $f(x)$ in the Eq. (16.39), the integral picks out the value that $\psi(x)$ takes on when $x$ is equal $0$; and, since the integral must be independent of $\psi(x)$ for all values of $x$ other than $0$, the function $\delta(x)$ must be $0$ everywhere except at $x=0$. Summarizing, we write \begin{equation} \label{Eq:III:16:40} \braket{0}{x}=\delta(x), \end{equation} where $\delta(x)$ is defined by \begin{equation} \label{Eq:III:16:41} \psi(0)=\int\delta(x)\psi(x)\,dx. \end{equation} Notice what happens if we use the special function “$1$” for the function $\psi$ in Eq. (16.41). Then we have the result \begin{equation} \label{Eq:III:16:42} 1=\int\delta(x)\,dx. \end{equation} That is, the function $\delta(x)$ has the property that it is $0$ everywhere except at $x=0$ but has a finite integral equal to unity. We must imagine that the function $\delta(x)$ has such a fantastic infinity at one point that the total area comes out equal to one.
One way of imagining what the Dirac $\delta$-function is like is to think of a sequence of rectangles—or any other peaked function you care to—which gets narrower and narrower and higher and higher, always keeping a unit area, as sketched in Fig. 16–2. The integral of this function from $-\infty$ to $+\infty$ is always $1$. If you multiply it by any function $\psi(x)$ and integrate the product, you get something which is approximately the value of the function at $x=0$, the approximation getting better and better as you use the narrower and narrower rectangles. You can if you wish, imagine the $\delta$-function in terms of this kind of limiting process. The only important thing, however, is that the $\delta$-function is defined so that Eq. (16.41) is true for every possible function $\psi(x)$. That uniquely defines the $\delta$-function. Its properties are then as we have described.
If we change the argument of the $\delta$-function from $x$ to $x-x'$, the corresponding relations are \begin{gather} \delta(x-x')=0,\quad x'\neq x,\notag\\[2ex] \label{Eq:III:16:43} \int\delta(x-x')\psi(x)\,dx=\psi(x'). \end{gather} If we use $\delta(x-x')$ for the amplitude $\braket{x}{x'}$ in Eq. (16.38), that equation is satisfied. Our result then is that for our base states in $x$, the condition corresponding to (16.36) is \begin{equation} \label{Eq:III:16:44} \braket{x'}{x}=\delta(x-x'). \end{equation}
We have now completed the necessary modifications of our basic equations which are necessary for dealing with the continuum of base states corresponding to the points along a line. The extension to three dimensions is fairly obvious; first we replace the coordinate $x$ by the vector $\FLPr$. Then integrals over $x$ become replaced by integrals over $x$, $y$, and $z$. In other words, they become volume integrals. Finally, the one-dimensional $\delta$-function must be replaced by just the product of three $\delta$-functions, one in $x$, one in $y$, and the other in $z$, $\delta(x-x')\,\delta(y-y')\,\delta(z-z')$. Putting everything together we get the following set of equations for the amplitudes for a particle in three dimensions: \begin{gather} \label{Eq:III:16:45} \braket{\phi}{\psi}=\int \braket{\phi}{\FLPr}\braket{\FLPr}{\psi}\, dV,\\[1.5ex] \label{Eq:III:16:46} \begin{aligned} \braket{\FLPr}{\psi}&=\psi(\FLPr),\\[1.5ex] \braket{\FLPr}{\phi}&=\phi(\FLPr), \end{aligned}\\[1.5ex] \label{Eq:III:16:47} \braket{\phi}{\psi}=\int \phi\cconj(\FLPr)\psi(\FLPr)\,dV,\\[1.5ex] \label{Eq:III:16:48} \braket{\FLPr'}{\FLPr}=\delta(x-x')\,\delta(y-y')\,\delta(z-z'). \end{gather}
What happens when there is more than one particle? We will tell you about how to handle two particles and you will easily see what you must do if you want to deal with a larger number. Suppose there are two particles, which we can call particle No. $1$ and particle No. $2$. What shall we use for the base states? One perfectly good set can be described by saying that particle $1$ is at $x_1$ and particle $2$ is at $x_2$, which we can write as $\ket{x_1,x_2}$. Notice that describing the position of only one particle does not define a base state. Each base state must define the condition of the entire system. You must not think that each particle moves independently as a wave in three dimensions. Any physical state $\ket{\psi}$ can be defined by giving all of the amplitudes $\braket{x_1,x_2}{\psi}$ to find the two particles at $x_1$ and $x_2$. This generalized amplitude is therefore a function of the two sets of coordinates $x_1$ and $x_2$. You see that such a function is not a wave in the sense of an oscillation that moves along in three dimensions. Neither is it generally simply a product of two individual waves, one for each particle. It is, in general, some kind of a wave in the six dimensions defined by $x_1$ and $x_2$. If there are two particles in nature which are interacting, there is no way of describing what happens to one of the particles by trying to write down a wave function for it alone. The famous paradoxes that we considered in earlier chapters—where the measurements made on one particle were claimed to be able to tell what was going to happen to another particle, or were able to destroy an interference—have caused people all sorts of trouble because they have tried to think of the wave function of one particle alone, rather than the correct wave function in the coordinates of both particles. The complete description can be given correctly only in terms of functions of the coordinates of both particles.
16–5The Schrödinger equation
So far we have just been worrying about how we can describe states which may involve an electron being anywhere at all in space. Now we have to worry about putting into our description the physics of what can happen in various circumstances. As before, we have to worry about how states can change with time. If we have a state $\ket{\psi}$ which goes over into another state $\ket{\psi'}$ sometime later, we can describe the situation for all times by making the wave function—which is just the amplitude $\braket{\FLPr}{\psi}$—a function of time as well as a function of the coordinate. A particle in a given situation can then be described by giving a time-varying wave function $\psi(\FLPr,t)=\psi(x,y,z,t)$. This time-varying wave function describes the evolution of successive states that occur as time develops. This so-called “coordinate representation”—which gives the projections of the state $\ket{\psi}$ into the base states $\ket{\FLPr}$ may not always be the most convenient one to use—but we will consider it first.
In Chapter 8 we described how states varied in time in terms of the Hamiltonian $H_{ij}$. We saw that the time variation of the various amplitudes was given in terms of the matrix equation \begin{equation} \label{Eq:III:16:49} i\hbar\,\ddt{C_i}{t}=\sum_jH_{ij}C_j. \end{equation} This equation says that the time variation of each amplitude $C_i$ is proportional to all of the other amplitudes $C_j$, with the coefficients $H_{ij}$.
How would we expect Eq. (16.49) to look when we are using the continuum of base states $\ket{x}$? Let’s first remember that Eq. (16.49) can also be written as \begin{equation*} i\hbar\,\ddt{}{t}\,\braket{i}{\psi}= \sum_j\bracket{i}{\Hop}{j}\braket{j}{\psi}. \end{equation*} Now it is clear what we should do. For the $x$-representation we would expect \begin{equation} \label{Eq:III:16:50} i\hbar\,\ddp{}{t}\,\braket{x}{\psi}= \int\bracket{x}{\Hop}{x'}\braket{x'}{\psi}\,dx'. \end{equation} The sum over the base states $\ket{j}$, gets replaced by an integral over $x'$. Since $\bracket{x}{\Hop}{x'}$ should be some function of $x$ and $x'$, we can write it as $H(x,x')$—which corresponds to $H_{ij}$ in Eq. (16.49). Then Eq. (16.50) is the same as \begin{gather} \label{Eq:III:16:51} i\hbar\,\ddp{}{t}\,\psi(x)=\int \kern -.6ex H(x,x')\psi(x')\,dx' \end{gather} with \begin{gather*} H(x,x')\equiv\bracket{x}{\Hop}{x'}. \end{gather*} According to Eq. (16.51), the rate of change of $\psi$ at $x$ would depend on the value of $\psi$ at all other points $x'$; the factor $H(x,x')$ is the amplitude per unit time that the electron will jump from $x'$ to $x$. It turns out in nature, however, that this amplitude is zero except for points $x'$ very close to $x$. This means—as we saw in the example of the chain of atoms at the beginning of the chapter, Eq. (16.12)—that the right-hand side of Eq. (16.51) can be expressed completely in terms of $\psi$ and the derivatives of $\psi$ with respect to $x$, all evaluated at the position $x$.
For a particle moving freely in space with no forces, no disturbances, the correct law of physics is \begin{equation*} \int\kern -.6ex H(x,x')\psi(x')\,dx'=-\frac{\hbar^2}{2m}\, \frac{\partial^2}{\partial x^2}\,\psi(x). \end{equation*} Where did we get that from? Nowhere. It’s not possible to derive it from anything you know. It came out of the mind of Schrödinger, invented in his struggle to find an understanding of the experimental observations of the real world. You can perhaps get some clue of why it should be that way by thinking of our derivation of Eq. (16.12) which came from looking at the propagation of an electron in a crystal.
Of course, free particles are not very exciting. What happens if we put forces on the particle? Well, if the force of a particle can be described in terms of a scalar potential $V(x)$—which means we are thinking of electric forces but not magnetic forces—and if we stick to low energies so that we can ignore complexities which come from relativistic motions, then the Hamiltonian which fits the real world gives \begin{equation} \label{Eq:III:16:52} \int\kern -.6ex H(x,x')\psi(x')\,dx'=-\frac{\hbar^2}{2m}\, \frac{\partial^2}{\partial x^2}\,\psi(x) +V(x)\psi(x). \end{equation} Again, you can get some clue as to the origin of this equation if you go back to the motion of an electron in a crystal, and see how the equations would have to be modified if the energy of the electron varied slowly from one atomic site to the other—as it might do if there were an electric field across the crystal. Then the term $E_0$ in Eq. (16.7) would vary slowly with position and would correspond to the new term we have added in (16.52).
[You may be wondering why we went straight from Eq. (16.51) to Eq. (16.52) instead of just giving you the correct function for the amplitude $H(x,x')=\bracket{x}{\Hop}{x'}$. We did that because $H(x,x')$ can only be written in terms of strange algebraic functions, although the whole integral on the right-hand side of Eq. (16.51) comes out in terms of things you are used to. If you are really curious, $H(x,x')$ can be written in the following way: \begin{equation*} H(x,x')=-\frac{\hbar^2}{2m}\,\delta''(x-x') +V(x)\,\delta(x-x'), \end{equation*} where $\delta''$ means the second derivative of the delta function. This rather strange function can be replaced by a somewhat more convenient algebraic differential operator, which is completely equivalent: \begin{equation*} H(x,x')=\biggl\{ -\frac{\hbar^2}{2m}\,\frac{\partial^2}{\partial x^2}+V(x)\biggr\} \delta(x-x'). \end{equation*} We will not be using these forms, but will work directly with the form in Eq. (16.52).]
If we now use the expression we have in (16.52) for the integral in (16.50) we get the following differential equation for $\psi(x)=\braket{x}{\psi}$: \begin{equation} \label{Eq:III:16:53} i\hbar\,\ddp{\psi}{t}=-\frac{\hbar^2}{2m}\, \frac{\partial^2}{\partial x^2}\,\psi(x)+V(x)\psi(x). \end{equation}
It is fairly obvious what we should use instead of Eq. (16.53) if we are interested in motion in three dimensions. The only changes are that $\partial^2/\partial x^2$ gets replaced by \begin{equation*} \nabla^2=\frac{\partial^2}{\partial x^2} +\frac{\partial^2}{\partial y^2} +\frac{\partial^2}{\partial z^2}, \end{equation*} and $V(x)$ gets replaced by $V(x,y,z)$. The amplitude $\psi(x,y,z)$ for an electron moving in a potential $V(x,y,z)$ obeys the differential equation \begin{equation} \label{Eq:III:16:54} i\hbar\,\ddp{\psi}{t}=-\frac{\hbar^2}{2m}\, \nabla^2\psi+V\psi. \end{equation} It is called the Schrödinger equation, and was the first quantum-mechanical equation ever known. It was written down by Schrödinger before any of the other quantum equations we have described in this book were discovered.
Although we have approached the subject along a completely different route, the great historical moment marking the birth of the quantum mechanical description of matter occurred when Schrödinger first wrote down his equation in 1926. For many years the internal atomic structure of matter had been a great mystery. No one had been able to understand what held matter together, why there was chemical binding, and especially how it could be that atoms could be stable. Although Bohr had been able to give a description of the internal motion of an electron in a hydrogen atom which seemed to explain the observed spectrum of light emitted by this atom, the reason that electrons moved in this way remained a mystery. Schrödinger’s discovery of the proper equations of motion for electrons on an atomic scale provided a theory from which atomic phenomena could be calculated quantitatively, accurately, and in detail. In principle, Schrödinger’s equation is capable of explaining all atomic phenomena except those involving magnetism and relativity. It explains the energy levels of an atom, and all the facts of chemical binding. This is, however, true only in principle—the mathematics soon becomes too complicated to solve exactly any but the simplest problems. Only the hydrogen and helium atoms have been calculated to a high accuracy. However, with various approximations, some fairly sloppy, many of the facts of more complicated atoms and of the chemical binding of molecules can be understood. We have shown you some of these approximations in earlier chapters.
The Schrödinger equation as we have written it does not take into account any magnetic effects. It is possible to take such effects into account in an approximate way by adding some more terms to the equation. However, as we have seen in Volume II, magnetism is essentially a relativistic effect, and so a correct description of the motion of an electron in an arbitrary electromagnetic field can only be discussed in a proper relativistic equation. The correct relativistic equation for the motion of an electron was discovered by Dirac a year after Schrödinger brought forth his equation, and takes on quite a different form. We will not be able to discuss it at all here.
Before we go on to look at some of the consequences of the Schrödinger equation, we would like to show you what it looks like for a system with a large number of particles. We will not be making any use of the equation, but just want to show it to you to emphasize that the wave function $\psi$ is not simply an ordinary wave in space, but is a function of many variables. If there are many particles, the equation becomes \begin{equation} \label{Eq:III:16:55} i\hbar\,\ddp{\psi(\FLPr_1,\FLPr_2,\FLPr_3,\dotsc)}{t}= \sum_i-\frac{\hbar^2}{2m_i}\biggl\{\! \frac{\partial^2\psi}{\partial x_i^2} +\frac{\partial^2\psi}{\partial y_i^2} +\frac{\partial^2\psi}{\partial z_i^2}\!\biggr\} +V(\FLPr_1,\FLPr_2,\dotsc)\psi. \end{equation} The potential function $V$ is what corresponds classically to the total potential energy of all the particles. If there are no external forces acting on the particles, the function $V$ is simply the electrostatic energy of interaction of all the particles. That is, if the $i$th particle carries the charge $Z_iq_e$, then the function $V$ is simply3 \begin{equation} \label{Eq:III:16:56} V(\FLPr_1,\FLPr_2,\FLPr_3,\dotsc)= \sum_{\substack{\text{all}\\\text{pairs}}} \frac{Z_iZ_j}{r_{ij}}\,e^2. \end{equation}
16–6Quantized energy levels
In a later chapter we will look in detail at a solution of Schrödinger’s equation for a particular example. We would like now, however, to show you how one of the most remarkable consequence of Schrödinger’s equation comes about—namely, the surprising fact that a differential equation involving only continuous functions of continuous variables in space can give rise to quantum effects such as the discrete energy levels in an atom. The essential fact to understand is how it can be that an electron which is confined to a certain region of space by some kind of a potential “well” must necessarily have only one or another of a certain well-defined set of discrete energies.
Suppose we think of an electron in a one-dimensional situation in which its potential energy varies with $x$ in a way described by the graph in Fig. 16–3. We will assume that this potential is static—it doesn’t vary with time. As we have done so many times before, we would like to look for solutions corresponding to states of definite energy, which means, of definite frequency. Let’s try a solution of the form \begin{equation} \label{Eq:III:16:57} \psi=a(x)e^{-iEt/\hbar}. \end{equation} If we substitute this function into the Schrödinger equation, we find that the function $a(x)$ must satisfy the following differential equation: \begin{equation} \label{Eq:III:16:58} \frac{d^2a(x)}{dx^2}=\frac{2m}{\hbar^2} [V(x)-E]a(x). \end{equation} This equation says that at each $x$ the second derivative of $a(x)$ with respect to $x$ is proportional to $a(x)$, the coefficient of proportionality being given by the quantity $(2m/\hbar^2)(V-E)$. The second derivative of $a(x)$ is the rate of change of its slope. If the potential $V$ is greater than the energy $E$ of the particle, the rate of change of the slope of $a(x)$ will have the same sign as $a(x)$. That means that the curve of $a(x)$ will be concave away from the $x$-axis. That is, it will have, more or less, the character of the positive or negative exponential function, $e^{\pm x}$. This means that in the region to the left of $x_1$, in Fig. 16–3, where $V$ is greater than the assumed energy $E$, the function $a(x)$ would have to look like one or another of the curves shown in part (a) of Fig. 16–4.
If, on the other hand, the potential function $V$ is less than the energy $E$, the second derivative of $a(x)$ with respect to $x$ has the opposite sign from $a(x)$ itself, and the curve of $a(x)$ will always be concave toward the $x$-axis like one of the pieces shown in part (b) of Fig. 16–4. The solution in such a region has, piece-by-piece, roughly the form of a sinusoidal curve.
Now let’s see if we can construct graphically a solution for the function $a(x)$ which corresponds to a particle of energy $E_a$ in the potential $V$ shown in Fig. 16–3. Since we are trying to describe a situation in which a particle is bound inside the potential well, we want to look for solutions in which the wave amplitude takes on very small values when $x$ is way outside the potential well. We can easily imagine a curve like the one shown in Fig. 16–5 which tends toward zero for large negative values of $x$, and grows smoothly as it approaches $x_1$. Since $V$ is equal to $E_a$ at $x_1$, the curvature of the function becomes zero at this point. Between $x_1$ and $x_2$, the quantity $V-E_a$ is always a negative number, so the function $a(x)$ is always concave toward the axis, and the curvature is larger the larger the difference between $E_a$ and $V$. If we continue the curve into the region between $x_1$ and $x_2$, it should go more or less as shown in Fig. 16–5.
Now let’s continue this curve into the region to the right of $x_2$. There it curves away from the axis and takes off toward large positive values, as drawn in Fig. 16–6. For the energy $E_a$ we have chosen, the solution for $a(x)$ gets larger and larger with increasing $x$. In fact, its curvature is also increasing (if the potential continues to stay flat). The amplitude rapidly grows to immense proportions. What does this mean? It simply means that the particle is not “bound” in the potential well. It is infinitely more likely to be found outside of the well, than inside. For the solution we have manufactured, the electron is more likely to be found at $x=+\infty$ than anywhere else. We have failed to find a solution for a bound particle.
Let’s try another energy, say one a little bit higher than $E_a$—say the energy $E_b$ in Fig. 16–7. If we start with the same conditions on the left, we get the solution drawn in the lower half of Fig. 16–7. It looked at first as though it were going to be better, but it ends up just as bad as the solution for $E_a$—except that now $a(x)$ is getting more and more negative as we go toward large values of $x$.
Maybe that’s the clue. Since changing the energy a little bit from $E_a$ to $E_b$ causes the curve to flip from one side of the axis to the other, perhaps there is some energy lying between $E_a$ and $E_b$ for which the curve will approach zero for large values of $x$. There is, indeed, and we have sketched how the solution might look in Fig. 16–8.
You should appreciate that the solution we have drawn in the figure is a very special one. If we were to raise or lower the energy ever so slightly, the function would go over into curves like one or the other of the two broken-line curves shown in Fig. 16–8, and we would not have the proper conditions for a bound particle. We have obtained a result that if a particle is to be bound in a potential well, it can do so only if it has a very definite energy.
Does that mean that there is only one energy for a particle bound in a potential well? No. Other energies are possible, but not energies too close to $E_c$. Notice that the wave function we have drawn in Fig. 16–8 crosses the axis four times in the region between $x_1$ and $x_2$. If we were to pick an energy quite a bit lower than $E_c$, we could have a solution which crosses the axis only three times, only two times, only once, or not at all. The possible solutions are sketched in Fig. 16–9. (There may also be other solutions corresponding to values of the energy higher than the ones shown.) Our conclusion is that if a particle is bound in a potential well, its energy can take on only the certain special values in a discrete energy spectrum. You see how a differential equation can describe the basic fact of quantum physics.
We might remark one other thing. If the energy $E$ is above the top of the potential well, then there are no longer any discrete solutions, and any possible energy is permitted. Such solutions correspond to the scattering of free particles by a potential well. We have seen an example of such solutions when we considered the effects of impurity atoms in a crystal.
- You can imagine that as the points $x_n$ get closer together, the amplitude $A$ to jump from $x_{n\pm1}$ to $x_n$ will increase. ↩
- For a discussion of probability distributions see Vol. I, Section 6–4. ↩
- We are using the convention of the earlier volumes according to which $e^2\equiv q_e^2/4\pi\epsO$. ↩